|

2023年12月16日,科学指南针主办的首届多尺度材料计算模拟国际研讨会在北京圆满落幕。来自国内外100多位专家学者共聚一堂,共同探讨材料计算领域的研究成果和发展方向,带来了一场精彩纷呈的学术盛宴。小编将采撷与会专家学者的精彩观点,以飨读者。
大会报告观点集锦
01
穆希皎
科学指南针高级顾问
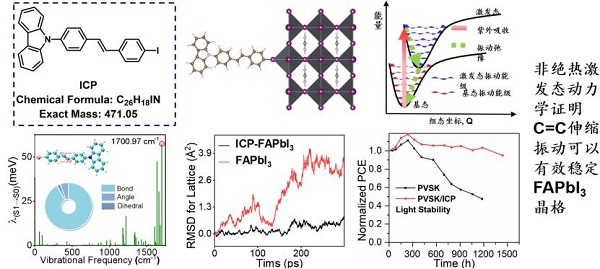
报告题目:超分子表界面光电热理论模拟
在光电热模拟计算领域,存在结构多样性、动态性和多尺度耦合等行业难点、痛点,亟待突破光/电/热过程协同效应、光/电/热多尺度相互作用、时间与精度的平衡等技术性瓶颈https://www.shiyanjia.com/。
具体来看,其中存在两类关键科学问题。一是表界面的结构、动态性与复杂交互,这主要体现在如何预测和理解表界面光电热的协同效应,以及其对物质构效关系的影响。二是多尺度耦合与人工智能在理论计算中的角色,主要包括第一性原理与机器学习技术融合的模拟预测等。基于此,该报告从极化子传输机制、表面类金属性、光-声转化稳定钙钛矿太阳能电池、人工智能与表界面传输机制等多个角度对两类科学问题进行了精彩回应,提出了基于光电热过程统一理论实现精准超分子理性设计的新策略。
02
马志远
西南石油大学副研究员
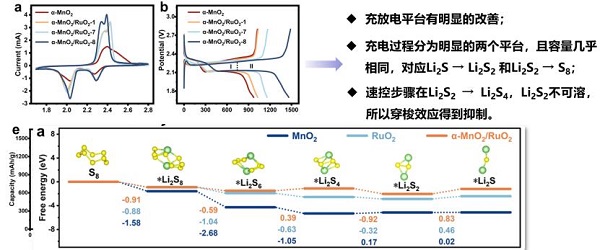
报告题目:锂硫电池多硫化物催化转化机制的DFT计算研究
在锂硫电池领域,长久以来存在多硫化物“穿梭效应”、锂枝晶生长以及硫和反应产物的绝缘性三大性能瓶颈,造成电芯能量密度降低、循环寿命短、稳定性差以及电芯安全管理难度大等现实问题。研究制备了一种新型二氧化钌纳米颗粒负载二氧化锰材料,二者形成特殊的共格界面结构。该材料导电性好、机械性能高、化学吸附强、催化活性高,是制作锂硫电池隔膜的理想材料。理论计算和实验研究表明该材料能够很好地稳定化溶解性锂硫化合物,为设计合适的正极材料以及隔膜材料提供支撑。
03
Younis Umer
中国电子科技大学研究员
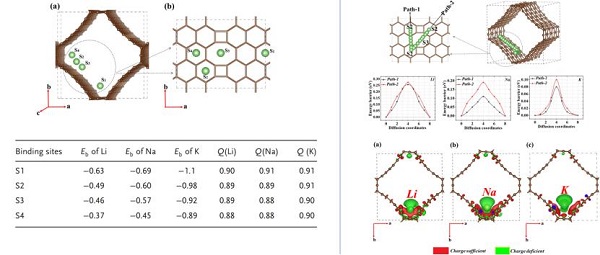
报告题目:Rational Design of 3D Porous Materials As Anode for Metal-Ion Batteries
长久以来,碳材料,尤其是石墨类材料被广泛用作锂离子电池负极材料。当前,钠离子电池、钾离子电池甚至钙离子电池相关工作不断涌现,但是传统的石墨类负极材料由于尺寸因素,不利于更大尺寸阳离子的嵌入与迁移,影响电池容量与安全性。针对新型碱金属和碱土金属离子电池设计新的负极材料成为关键科学问题。将电池结构负极孔道结构从二维扩展为三维结构无疑是非常新颖的思路。当前三维多孔模型正在成为负极材料领域冉冉升起的新星。Umer研究员以第一性原理计算为手段,对周期性三维多孔碳、三维多孔碳化磷硼、三维多孔硼氮化硅以及三维磷掺杂石墨炔材料的电子结构性能,碱金属离子的结合以及迁移行为等性质进行深入细致的研究。上述这些材料的导电性能从金属性、半金属性到半导体性质各不相同,均具有比容量高、离子迁移能垒低(大体积碱金属)等突出优势,适应于新型电池多种应用场景。
04
唐法威
科学指南针高级计算工程师
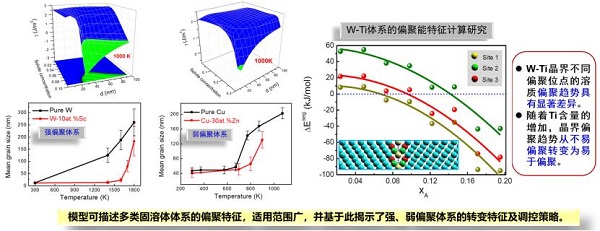
报告题目:金属基纳米结构高温稳定化的理论计算研究
金属基纳米结构材料在民用和军工领域均具有广阔的应用前景,尤其对于开发可在极端服役环境下保持优异综合力学性能和理化特性的金属基材料体系具有重要战略意义。其中的关键工程问题是复杂环境下的体系高温稳定化,具体包括晶粒组织高温稳定性和物相构成高温稳定性。
总体来看,需要将合金体系成分设计、纳米结构合成制备、组织结构高温调控三方面相互结合。因此,该报告从高温稳定化的基础科学问题出发,针对固溶体体系、相分离体系和不互溶体系进行了分类研究,构建了二元纳米晶合金体系高普适度、多尺度、物理意义明确的热力学模型,采用理论与实验相结合的方式,提出以偏聚效应为主导的高温稳定化调控机制,对开发具有高热/相稳定性的金属基纳米材料提供了新的设计原则。
05
盛翔
中国科学院天津工业生物技术研究所研究员
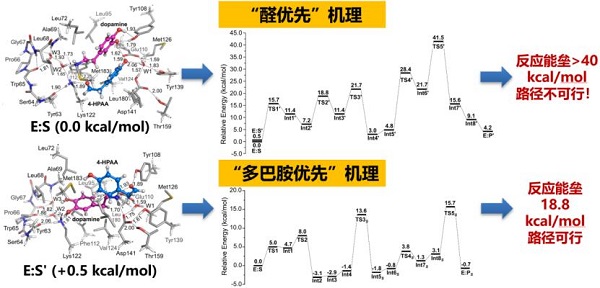
报告题目:酶的计算模拟与理性设计
以生物催化剂为核心的工业生物技术被成为生物技术发展的“第三次浪潮”。通过结合量子化学、分子动力学和人工智能技术,实现对酶促进反应机理、蛋白结构动态变化等过程的深入理解和调控,从而可以指导人工酶的设计开发,在天然酶的基础上进行结构改造,以适应工业过程的需要。为了实现这一目标,需要计算与实验工作者充分协作、互相理解。在报告中,介绍了相关研究经历,包括通过计算纠正了被错误解析的蛋白质晶体、通过QM/MM方法揭示了酶促反应机理、并进一步对酶进行理性改造以实现手性调控等方面的进展。
06
刘昭睿
北京航空航天大学博士研究生
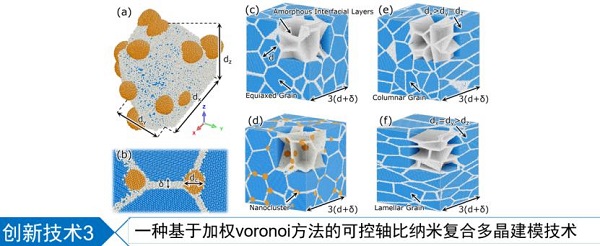
报告题目:原子级材料建模、模拟、分析和表征的高通量集成化平台:SPaMD
计算驱动与数据驱动构成了新一代材料开发方法,为了实现这一目标篇需要自动化、集成化、模块化的高通量计算,SPaMD正是为这一目的而开发的建模-计算-后处理-数据分析集成化平台。SPaMD可调用多种主流第一性原理计算程序和自行开发的计算内核,支持强大的建模功能,包括晶界、多晶、孪晶、异质结自动建模、吸附位点自动搜索等。在计算结果方面,可以进行丰富的可视化和高级分析。除第一性原理外,对于分子动力学等应用于大尺度体系的计算也有强大的支持。
07
王超
天津城建大学讲师
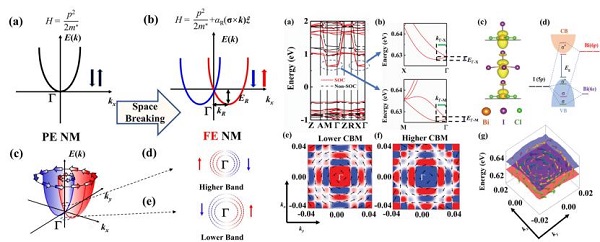
报告题目:低维有机无机杂化铁电钙钛矿中物性的第一性原理研究
有机无机杂化钙钛矿材料是太阳能电池领域的研究重点之一。为了获得具有更高稳定性、高铁电极化、高光吸收载流子迁移和非铅元素的太阳能电池材料体系,新型高性能杂化钙钛矿材料亟待开发。
微观尺度计算对于材料结构和成分的设计具有极大的优势,可以有效缩短材料研发周期、降低研发成本。基于此,该报告通过第一性原理计算揭示了无机八面体位移极化对于总极化起到的关键作用。在形变势理论基础下,发现(MV)BiI3Cl2和(MV)SbI3Cl2具有高的电子迁移率。在自旋-轨道耦合作用下,发现Rashba型自旋织构位置关系,Rashba常数随着极化值和拉伸的变化规律。计算系统阐述了有机无机杂合钙钛矿的性能与电子结构特征之间的关系,为研发新型高性能杂合钙钛矿材料提供了理论支撑。
08
焦东旭
吉林大学博士研究生
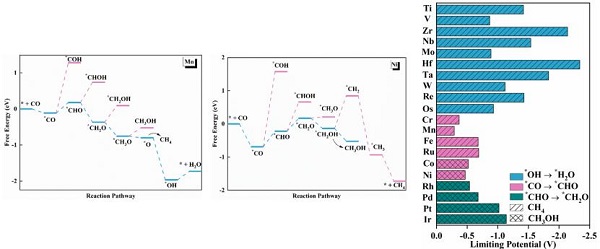
报告题目:过渡金属单原子负载N掺杂γ相石墨炔用于COER的理论研究
一氧化碳的界面还原机制研究一直是工业界前沿研究领域的重要课题。利用单原子的特殊电子结构改良CO的还原路径,实现高附加值化学品的高选择性生成是符合绿色化学理念并能够创造经济效益的重要途经。
研究采用氮掺杂石墨炔负载一系列过渡金属单原子结构,通过理论计算系统性比较这些过渡金属单原子的催化性能。研究发现不同过渡金属元素催化CO还原的决速步骤不同导致产物差异。前过渡金属更容易生成甲烷,后过渡金属容易生成甲醇。后过渡元素反应的决速步骤是*CO转化为*CHO中间体,理论过电位最低。该研究为进一步设计优良CO还原催化剂提供理论支撑。
09
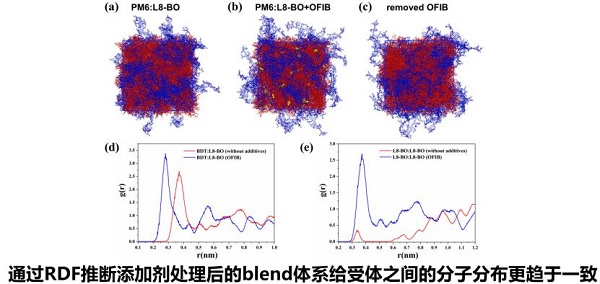
梁泽洲
西安交通大学博士研究生
报告题目:密度泛函理论和分子动力学模拟在有机光伏研究中的应用
有机太阳能电池可与硅电池互补,应用于柔性可穿戴便携式设备、光伏器件、国防军工等领域,具有重要研究价值。其中给受体材料的分子结构、分子间电荷转移机制以及分子堆积对器件性能的影响机制难以仅通过实验手段确认,需要通过理论计算的方式获得更深入的理解https://www.shiyanjia.com/。
基于此,该报告从有机光伏材料的激发态性质计算以及给受体界面匹配性、分子动力学模拟添加剂分子与给受体材料相互作用机制两方面展开论述,发现PM6:Y6分子间具有更好的分子堆积情况,π-π作用区域大,更有利于分子间电荷转移,PM6Y6之间的电子和空穴转移仅通过特殊的传输通道转移且电荷转移通道固定。系统研究了薄膜堆积状态与光谱之间的变化规律,分子堆积距离的微小变化显著影响电荷转移积分,进而影响分子间电荷转移。该研究对新型太阳能电池的设计研发提供了新的理论依据。
写在最后
本次研讨会精彩纷呈,9位计算领域的专家学者从理论构建、方法发展、集成平台的搭建等维度,深入探讨催化、电池、金属、铁电等多个材料方向的研究热点和产业进展,获得了观众的高度认可。未来科学指南针将在科研领域持续深耕,汇聚更多科学领域的专家智库资源,为科研工作者带来更专业、更高质量的学术交流活动。
|